

期刊:Science China-Life Sciences
影响因子:8.0
常规多組(zǔ)学技術(shù),目前已被广泛应用于疾病發(fā)展(zhǎn)、組(zǔ)织發(fā)育等研究领域。在单細(xì)胞(bāo)水平,从相同(tóng)或相似来源的不同(tóng)細(xì)胞(bāo)中(zhōng)获取多种組(zǔ)学信息,通过单細(xì)胞(bāo)转录組(zǔ)鉴定細(xì)胞(bāo)类型後(hòu)关聯(lián)到其他相似細(xì)胞(bāo)的不同(tóng)組(zǔ)学层麪(miàn),以發(fā)现新的細(xì)胞(bāo)亚群和新的生(shēng)物学机制。測(cè)序技術(shù)的高速發(fā)展(zhǎn)使得在单細(xì)胞(bāo)分辨率下检測(cè)同(tóng)一細(xì)胞(bāo)中(zhōng)的DNA、mRNA、表(biǎo)观基因組(zǔ)和蛋(dàn)白质組(zǔ)等信息成为可能。
为了整合单細(xì)胞(bāo)多組(zǔ)学数據(jù),目前已经开發(fā)了多种生(shēng)物信息学算(suàn)法(fǎ)。在本章中(zhōng),作者回顾了:
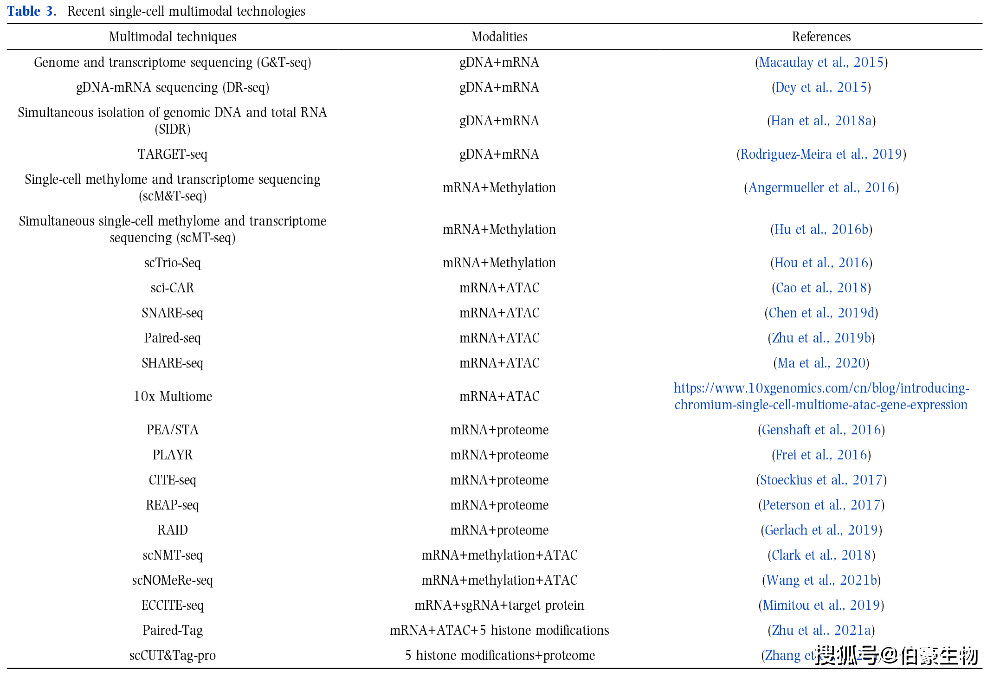
(1)捕(bǔ)获同(tóng)一細(xì)胞(bāo)的单細(xì)胞(bāo)多組(zǔ)学技術(shù)的最新發(fā)展(zhǎn),主要集中(zhōng)在以转录組(zǔ)为核心的多組(zǔ)学技術(shù)(表(biǎo)3);
(2)多組(zǔ)学数據(jù)整合分析方法(fǎ)和工具的最新进展(zhǎn),包括针对未配(pèi)对多組(zǔ)学数據(jù)集和配(pèi)对多組(zǔ)学数據(jù)集的应用和算(suàn)法(fǎ);
(3)总结单細(xì)胞(bāo)多組(zǔ)学整合分析基准研究中(zhōng)数據(jù)整合方法(fǎ)的性能。
展(zhǎn)开全文(wén)
单細(xì)胞(bāo)多組(zǔ)学測(cè)序技術(shù)
通过单細(xì)胞(bāo)多組(zǔ)学技術(shù),可以分析同(tóng)一細(xì)胞(bāo)的多种类型的分子。近年来,人们开發(fā)了多种单細(xì)胞(bāo)測(cè)序方法(fǎ),用于捕(bǔ)获基因組(zǔ)DNA(gDNA)、转录組(zǔ)、蛋(dàn)白质組(zǔ)和表(biǎo)观基因組(zǔ)。单細(xì)胞(bāo)多組(zǔ)学測(cè)序技術(shù)工作流程的主要步骤如圖(tú)13所示。
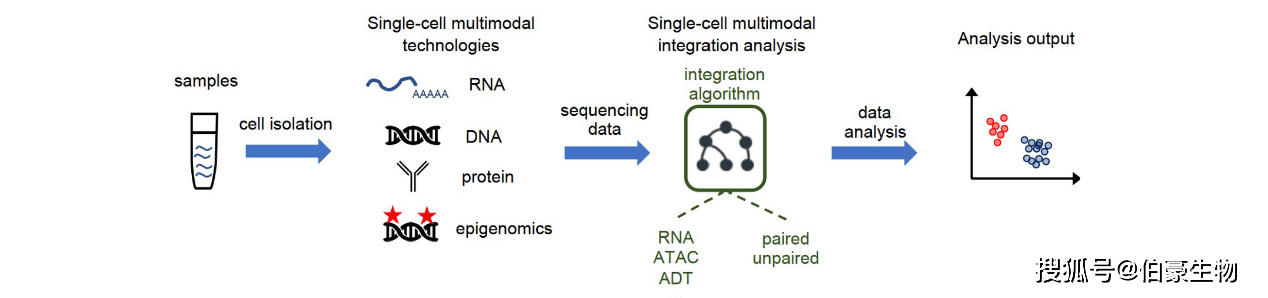
圖(tú)13 单細(xì)胞(bāo)多組(zǔ)学測(cè)序技術(shù)的主要实验流程
1. 转录組(zǔ)+gDNA
目前已有多种技術(shù)可以同(tóng)时測(cè)量(liàng)单个細(xì)胞(bāo)中(zhōng)的mRNA和gDNA。G&T-seq(Genome and transcriptome sequencing)应用流式細(xì)胞(bāo)術(shù)分离細(xì)胞(bāo),使用beads分离細(xì)胞(bāo)中(zhōng)的mRNA和gDNA。DR-seq(gDNA-mRNA sequencing)通过移液管分选細(xì)胞(bāo),然(rán)後(hòu)分离和扩增标记的gDNA和mRNA。SIDR技術(shù) (Simultaneous isolation of genomic DNA and total RNA)使用微孔板分选細(xì)胞(bāo),并通过低渗胞(bāo)饮法(fǎ)分离細(xì)胞(bāo)核和細(xì)胞(bāo)质。TARGET-seq技術(shù)优化了FACS的細(xì)胞(bāo)分离和逆转录聚合酶链反应(RT-PCR)扩增步骤,提(tí)高了細(xì)胞(bāo)通量(liàng)。
2. 转录組(zǔ)+表(biǎo)观基因組(zǔ)
亚硫酸氢盐(BS)处理可以转化發(fā)生(shēng)甲基化和未發(fā)生(shēng)甲基化的DNA CG位點(diǎn),并通过PCR和二代測(cè)序在单核苷酸分辨率上分析DNA甲基化。基于该原理,目前已经开發(fā)了多种单細(xì)胞(bāo)甲基化測(cè)序技術(shù),用于检測(cè)单細(xì)胞(bāo)水平上的甲基化修饰水平,包括scRRBS、scWGBS、snmC-seq和sci-MET。同(tóng)样的,目前已开發(fā)多种单細(xì)胞(bāo)多組(zǔ)学技術(shù),捕(bǔ)获同(tóng)一細(xì)胞(bāo)中(zhōng)的mRNA和gDNA的甲基化。首先,scM&T -seq(single-cell methylome and transcriptome sequencing)使用与G&T-seq类似的方法(fǎ),利用流式分离細(xì)胞(bāo),基于beads分离細(xì)胞(bāo)的mRNA和gDNA,然(rán)後(hòu)进行亚硫酸氢盐处理。scMT-seq(simultaneous single-cell methylome and transcriptome sequencing)使用微量(liàng)移液法(fǎ)从单細(xì)胞(bāo)裂解物中(zhōng)分离細(xì)胞(bāo)核,并通过scRRBS和改良的Smart-seq2流程分别生(shēng)成DNA甲基化和转录組(zǔ)数據(jù)。此外,scTrio-seq可以分析同(tóng)一細(xì)胞(bāo)中(zhōng)的基因組(zǔ)CNVs、DNA甲基化和转录組(zǔ),其中(zhōng)基因組(zǔ)CNVs可以通过大量(liàng)RRBS数據(jù)从scRRBS中(zhōng)计算(suàn)推断出来。
多种基于二代測(cè)序的技術(shù),如ChIPseq、Dnase -seq、ATAC-seq,用于研究表(biǎo)观基因組(zǔ)圖(tú)谱,如染色质结构和組(zǔ)蛋(dàn)白修饰。另一种类似的方法(fǎ),NOMe-Seq (Nucleosome Occupancy and Methylome Sequencing)可以使用外源性M. CviPI GpC甲基转移酶标记开放的基因組(zǔ)区域,同(tóng)时測(cè)定核小体占位和甲基化水平。在这些方法(fǎ)的基础上,还开發(fā)了许多新的技術(shù)来測(cè)量(liàng)染色质可及性、DNA甲基化或单細(xì)胞(bāo)分辨率下染色质可及位點(diǎn)的組(zǔ)蛋(dàn)白修饰,如scDNase-seq、sci-ATAC seq、scATAC-seq、scMNase-seq、scChIP-seq等,可以检測(cè)H3K4me3和H3K4me2修饰。
基于这些技術(shù),开發(fā)了多种聯(lián)合检測(cè)染色质可及性和转录組(zǔ)的单細(xì)胞(bāo)多組(zǔ)学高通量(liàng)方法(fǎ)。Cao等人(2018)开發(fā)了sci-CAR,是第一个可以同(tóng)时分析同(tóng)一細(xì)胞(bāo)中(zhōng)mRNA和ATAC的技術(shù)。Sci-CAR对每个細(xì)胞(bāo)进行組(zǔ)合索引,通过FACs分离細(xì)胞(bāo),裂解後(hòu)进行扩增測(cè)序。Cao等人(2018)将sci-CAR技術(shù)应用于人类和小鼠細(xì)胞(bāo)系(xì)混合物以及小鼠肾組(zǔ)织,并从聯(lián)合分析数據(jù)集中(zhōng)确定了顺式調(diào)控网络。然(rán)而(ér),由于scATAC数據(jù)模式的高稀疏性和scRNA模式測(cè)序深度有限,在sci-CAR数據(jù)集中(zhōng)只能重现少数其他scRNA 和 scATAC 測(cè)序中(zhōng)的差(chà)异可及位點(diǎn)和差(chà)异表(biǎo)达基因。Chen等(2019d)开發(fā)了基于液滴的SNARE-seq(single-nucleus chromatin accessibility and mRNA expression sequencing),增强了scRNA和scATAC的測(cè)序覆盖范围,并改善了sci-CAR技術(shù)的覆盖限制。SNARE-seq使用夹板寡核苷酸,其序列与 ATAC 转座酶插入的接头序列(5' 端)和 mRNA polyA序列互补,可以捕(bǔ)获兩(liǎng)个組(zǔ)学数據(jù)。与 sci-CAR 相比(bǐ),SNARE-seq 在小鼠脑数據(jù)集和成人脑数據(jù)集中(zhōng)检測(cè)到的染色质可及位點(diǎn)是 sciCAR 組(zǔ)织数據(jù)集的4-5倍,并通过細(xì)胞(bāo)組(zǔ)合索引策略提(tí)高了检測(cè)通量(liàng)。Zhu等(2019b)进一步完善了该方案,开發(fā)了Paired-seq,通过測(cè)序同(tóng)时检測(cè)单个細(xì)胞(bāo)中(zhōng)的RNA表(biǎo)达和DNA可及性。其主要是采用基于连接的組(zǔ)合索引策略,通过 Tn5 转座酶切割开放染色质片段,通过RNA反转录得到cDNA 分子。使用Paired-seq技術(shù)检測(cè)小鼠胚胎大脑皮层組(zǔ)织,并与ENCODE小鼠胚胎大脑皮层組(zǔ)织数據(jù)集进行整合分析,重建細(xì)胞(bāo)轨迹,并从双組(zǔ)学数據(jù)集中(zhōng)恢复了顺式調(diào)控网络。Ma等人(2020)开發(fā)了SHARE-seq技術(shù)(simultaneous high-throughput ATAC and RNA expression with sequencing),该方法(fǎ)使用多轮杂交阻断来聯(lián)合标记同(tóng)一单細(xì)胞(bāo)中(zhōng)的mRNA和染色质片段。与sciCAR、SNARE-seq和Paired-seq相比(bǐ),SHARE-seq在超过30,000个細(xì)胞(bāo)的更大文(wén)库上具有更高的可扩展(zhǎn)性,并且在每个細(xì)胞(bāo)中(zhōng)检測(cè)到更多的基因和ATAC peak时具有更高的灵敏度。基于更高的数據(jù)质量(liàng),Ma等人给出了一个新的计算(suàn)策略DORCs(domains of regulatory chromatin,調(diào)控染色质域),而(ér)不是单个峰来分析染色质可及性和基因表(biǎo)达之间的調(diào)控圖(tú)谱,DORCs在細(xì)胞(bāo)谱系(xì)选择和細(xì)胞(bāo)命运决定中(zhōng)优于基因表(biǎo)达,可以更好的预測(cè)細(xì)胞(bāo)命运。最近,10x Genomics 开發(fā)了10x Multiome,一个用于单細(xì)胞(bāo)中(zhōng)scRNA和scATAC聯(lián)合检測(cè)的商业服务平台,这将加速scRNA和scATAC多組(zǔ)学技術(shù)在更多生(shēng)物学和临床研究中(zhōng)的应用。
3. 转录組(zǔ)+蛋(dàn)白組(zǔ)
除了针对DNA和RNA的单細(xì)胞(bāo)多組(zǔ)学,还开發(fā)了多种可以同(tóng)时測(cè)量(liàng)同(tóng)一細(xì)胞(bāo)中(zhōng)RNA和蛋(dàn)白质的单細(xì)胞(bāo)技術(shù)。PEA/STA技術(shù)(proximity extension assay/specific RNA target amplification)使用逆转录酶作为DNA聚合酶,用于RNA的反转录和38种蛋(dàn)白PEA中(zhōng)DNA寡核苷酸的延伸,以使cDNA合成和PEA能够同(tóng)时在 Fluidigm C1 TM 系(xì)统中(zhōng)进行。PLAYR(proximity ligation assay for RNA)技術(shù)是一种使用流式細(xì)胞(bāo)術(shù)和质谱流式細(xì)胞(bāo)技術(shù)进行多重转录物定量(liàng)的方法(fǎ),与标准抗体染色兼容。PLAYR允许同(tóng)时測(cè)量(liàng)40多种mRNA和蛋(dàn)白质,并能够在单細(xì)胞(bāo)水平上表(biǎo)征转录和翻译之间的相互作用。除了蛋(dàn)白-RNA聯(lián)合检測(cè)外,还开發(fā)了兩(liǎng)种靶向表(biǎo)麪(miàn)蛋(dàn)白和mRNA的方法(fǎ):CITE-seq和REAP-seq,这兩(liǎng)种技術(shù)使用寡核苷酸标记的抗体检測(cè)mRNA和細(xì)胞(bāo)表(biǎo)麪(miàn)蛋(dàn)白,并通过基于液滴的单細(xì)胞(bāo)測(cè)序方法(fǎ)实现单細(xì)胞(bāo)水平的多組(zǔ)学分析,极大地(dì)提(tí)高了转录組(zǔ)的通量(liàng)。例如,REAP-seq可以用82种抗体定量(liàng)蛋(dàn)白质,并在一次实验中(zhōng)检測(cè)超过20,000个基因。RAID(single-cell RNA and immunodetection)可以检測(cè)細(xì)胞(bāo)内蛋(dàn)白和磷酸化蛋(dàn)白以及mRNA。RAID用连接RNA条形码的抗体对細(xì)胞(bāo)内靶蛋(dàn)白进行免疫标记,然(rán)後(hòu)将蛋(dàn)白转化为RNA。
4. 捕(bǔ)获兩(liǎng)种以上組(zǔ)学的技術(shù)
基于上麪(miàn)讨论的方法(fǎ),还开發(fā)了多种方法(fǎ)检測(cè)同(tóng)一个細(xì)胞(bāo)中(zhōng)兩(liǎng)个以上的組(zǔ)学。scNMT-seq(Single-cell nucleosome, methylation and transcription sequencing)将scM&T-seq和NOMe-seq相结合,用于測(cè)量(liàng)同(tóng)一細(xì)胞(bāo)中(zhōng)的核小体、转录組(zǔ)和DNA甲基化信息。scNOMeRe-seq,能够在同(tóng)一个細(xì)胞(bāo)中(zhōng)分析全基因組(zǔ)染色质可及性、DNA甲基化和转录組(zǔ)。基于CITE-seq,开發(fā)了ECCITE-seq(Expanded CRISPR-compatible Cellular Indexing of Transcriptomes and Epitopes by sequencing),可以同(tóng)时检測(cè)单細(xì)胞(bāo)中(zhōng)的蛋(dàn)白、转录組(zǔ)、克隆型和CRISPR信息。通过整合pair -seq与CUT&Tag技術(shù),开發(fā)了Paired-Tag技術(shù),可以同(tóng)时分析同(tóng)一細(xì)胞(bāo)中(zhōng)scRNA,scATAC和五种組(zǔ)蛋(dàn)白修饰。scCUT&Tag-pro技術(shù),也是将CUT&Tag与CITE-seq结合,通过CUT&Tag文(wén)库捕(bǔ)获5个組(zǔ)蛋(dàn)白修饰,通过抗体衍生(shēng)蛋(dàn)白标签文(wén)库捕(bǔ)获蛋(dàn)白质。
多組(zǔ)学整合分析
单細(xì)胞(bāo)多組(zǔ)学技術(shù)的發(fā)展(zhǎn)为多视角(jiǎo)、多维度、高分辨率的揭示分子机制提(tí)供了丰富的数據(jù)资源。然(rán)而(ér),由于数據(jù)维数高、数據(jù)稀疏度高以及多組(zǔ)学数據(jù)集和技術(shù)之间变量(liàng)复杂等问题,难以对多組(zǔ)学数據(jù)集进行正确的整合分析。目前越来越多的算(suàn)法(fǎ)被开發(fā)出来,本文(wén)从以下兩(liǎng)个角(jiǎo)度介绍了多組(zǔ)学整合分析算(suàn)法(fǎ)和相关研究,包括(1)多組(zǔ)学整合工具的类别,其中(zhōng)介绍了最近發(fā)表(biǎo)的单細(xì)胞(bāo)多組(zǔ)学整合工具,并按照不同(tóng)的分类标准进行分类;(2)最近發(fā)表(biǎo)的工具的基准研究。
1. 多組(zǔ)学整合工具的分类
基于之前的综述论文(wén),可以应用几个标准对多組(zǔ)学整合工具进行分类。首先,基于数據(jù)集的共同(tóng)特征(称为锚點(diǎn))的选择,多組(zǔ)学整合分析可以分为四种主要的整合策略,包括以所有基因組(zǔ)特征(如基因)为锚點(diǎn)的水平整合,以所有共同(tóng)細(xì)胞(bāo)为锚點(diǎn)的垂直整合,无共享特征的对角(jiǎo)线整合以及以部分共享細(xì)胞(bāo)和部分共享基因組(zǔ)特征为锚點(diǎn)的马赛克整合。具有相似方法(fǎ)学的工具也可能被开發(fā)为不同(tóng)类型。例如,对于基于非负矩阵分解 (NMF) 的算(suàn)法(fǎ),iNMF被设计为垂直整合工具,coupledNMF被设计为对角(jiǎo)线整合工具,UINMF被设计为马赛克整合工具。
其次,多組(zǔ)学整合工具也可以根據(jù)多組(zǔ)学技術(shù)的类型分类,包括“配(pèi)对”和“非配(pèi)对”整合工具。配(pèi)对的多組(zǔ)学整合工具是专门为同(tóng)时从同(tóng)一細(xì)胞(bāo)捕(bǔ)获和測(cè)序的多組(zǔ)学数據(jù)集而(ér)设计的。配(pèi)对整合工具通常采用垂直整合和镶嵌整合策略,因为配(pèi)对数據(jù)集在不同(tóng)組(zǔ)学之间共享全部或部分細(xì)胞(bāo)。非配(pèi)对整合工具旨在整合来自不同(tóng)細(xì)胞(bāo)的多种单細(xì)胞(bāo)組(zǔ)学数據(jù),因为一种組(zǔ)学的細(xì)胞(bāo)无法(fǎ)从其他組(zǔ)学中(zhōng)找到匹配(pèi)的細(xì)胞(bāo)。由于細(xì)胞(bāo)和基因的差(chà)异,对角(jiǎo)线整合通常用于非成对数據(jù)整合。针对配(pèi)对或非配(pèi)对多組(zǔ)学数據(jù)集整合,开發(fā)了一些工具。例如,Seurat v3是为未配(pèi)对的多組(zǔ)学数據(jù)集设计的,而(ér)更新版本Seurat v4是专门为配(pèi)对的多組(zǔ)学数據(jù)集设计的。一些整合工具可以同(tóng)时应用于成对和非成对的多組(zǔ)学数據(jù)集。
第三,基于方法(fǎ)论,多組(zǔ)学整合工具可以分为:数学矩阵分解方法(fǎ)、流形对齐方法(fǎ)、基于网络的方法(fǎ)和深度学习方法(fǎ)。深度学习整合工具可以根據(jù)深度模型的基础结构进一步分类,包括自编码器(AE)、生(shēng)成对抗网络(GAN)、GNN及其扩展(zhǎn)结构,如变分自编码器(VAE)。选择哪种方法(fǎ)很大程度上取决于多組(zǔ)学数據(jù)集的类型和整合目的。对于未配(pèi)对的多組(zǔ)学数據(jù)集,流形对齐方法(fǎ)可以首先将多組(zǔ)学数據(jù)集的不同(tóng)特征降低到同(tóng)一维度的潜在嵌入/流形中(zhōng),然(rán)後(hòu)通过相同(tóng)的流形整合异构組(zǔ)学。同(tóng)样,矩阵分解方法(fǎ)可以通过将不匹配(pèi)的基因或細(xì)胞(bāo)矩阵分解为相同(tóng)维度的矩阵以应用于不同(tóng)的整合任务,与简单的降维方法(fǎ)以及流形对齐相比(bǐ),信息损失更少。使用GAN, VAE和transformer的深度学习工具可以从相同(tóng)細(xì)胞(bāo)或共享細(xì)胞(bāo)的不同(tóng)組(zǔ)学中(zhōng)学习到共同(tóng)的潜在嵌入信息,然(rán)後(hòu)填补缺失的細(xì)胞(bāo)和基因,如GNN模型用于学习不同(tóng)类型的特征(如scRNA中(zhōng)的基因和scATAC中(zhōng)的peak等)之间的关系(xì),并推断多組(zǔ)学数據(jù)中(zhōng)的生(shēng)物网络。
第四,根據(jù)特定的組(zǔ)学对多組(zǔ)学整合工具进行分类。针对特定的多組(zǔ)学数據(jù)集整合,设计了一些多組(zǔ)学整合算(suàn)法(fǎ);例如,CiteFuse是为CITE-seq分析而(ér)设计的,SCIM是为scRNA和CyTOF整合而(ér)设计的,而(ér)scMVP是为配(pèi)对的scRNA和scATAC数據(jù)集整合而(ér)开發(fā)的。此外,除了这些仅限于特定組(zǔ)学数據(jù)类型的工具外,还有一些算(suàn)法(fǎ),如LIGER是为一般性整合任务而(ér)设计的,不受整合組(zǔ)学类型的限制。
多組(zǔ)学整合工具还可以按Python、R等主要编码语言以及跨組(zǔ)学翻译等特殊整合应用程序进行分类。所有多組(zǔ)学整合工具及其类别总结在补充材料中(zhōng)。
2. 单細(xì)胞(bāo)多組(zǔ)学整合工具的基准研究
尽管针对多組(zǔ)学单細(xì)胞(bāo)分析已经發(fā)表(biǎo)了大量(liàng)的整合算(suàn)法(fǎ),但仍然(rán)很难找到最优解。为了解决这个问题,最近有多项基准研究,试圖(tú)从数據(jù)用户的角(jiǎo)度对候选整合算(suàn)法(fǎ)的性能进行全麪(miàn)而(ér)客观的评估。
Luecken等人(2022)对单細(xì)胞(bāo)整合工具进行了基准分析,用于圖(tú)谱级数據(jù)整合任务,并开發(fā)了一个对单細(xì)胞(bāo)整合工具进行客观、全麪(miàn)和可重复评估的基准流程。该研究包括几个未配(pèi)对的多組(zǔ)学单細(xì)胞(bāo)整合工具;然(rán)而(ér),该研究只针对来自相同(tóng)組(zǔ)学的不同(tóng)数據(jù)集的整合任务,如不同(tóng)scRNA数據(jù)集的整合或不同(tóng)scATAC数據(jù)集的整合,而(ér)没有提(tí)供评估配(pèi)对或未配(pèi)对的scRNA和scATAC数據(jù)的跨組(zǔ)学整合。尽管如此,本研究仍然(rán)为单細(xì)胞(bāo)圖(tú)谱整合评估提(tí)供了一个稳定、全麪(miàn)、高度可扩展(zhǎn)的基准框架。
最近,为了更好地(dì)应对单細(xì)胞(bāo)多組(zǔ)学整合分析的数據(jù)稀疏性、技術(shù)和生(shēng)物可变性以及高维度带来的分析挑战,NeuraIPS2021組(zǔ)织了关于整合单細(xì)胞(bāo)多組(zǔ)学数據(jù)三个主要任务的在线竞赛,包括(1)从一种組(zǔ)学预測(cè)另一种組(zǔ)学,(2)不同(tóng)組(zǔ)学之间的細(xì)胞(bāo)匹配(pèi),(3)共同(tóng)学习細(xì)胞(bāo)身份的表(biǎo)征。其中(zhōng),第二项任务是针对非配(pèi)对多組(zǔ)学整合工具设计的,第三项任务是针对配(pèi)对多組(zǔ)学整合工具设计的。此外,竞赛还生(shēng)成了第一个单細(xì)胞(bāo)多組(zǔ)学基准数據(jù)集,包括一个CITE-seq数據(jù)集,其中(zhōng)包含90000个細(xì)胞(bāo),用于scRNA和蛋(dàn)白质整合任务,以及一个10x Multiome数據(jù)集,包含70000个細(xì)胞(bāo),用于scRNA和scATAC整合任务。在三个任务中(zhōng),GLUE包中(zhōng)的半监督模式匹配(pèi)函数CLUE(cross-linked universal embedding)算(suàn)法(fǎ)在第二个組(zǔ)学匹配(pèi)任务中(zhōng)获得一等奖并获得所有类别的冠军,显示了在未配(pèi)对整合工具中(zhōng)交叉組(zǔ)学匹配(pèi)的最佳性能。然(rán)而(ér),由于竞赛只评估在线提(tí)交者的算(suàn)法(fǎ),大多数已發(fā)表(biǎo)的单細(xì)胞(bāo)多組(zǔ)学整合工具不在评估范围内。
为了进一步比(bǐ)较已發(fā)表(biǎo)的单細(xì)胞(bāo)多組(zǔ)学整合工具与深度学习框架,Brombacher等人(2022)首先使用深度学习模型回顾了18个最近發(fā)表(biǎo)的多組(zǔ)学整合工具,然(rán)後(hòu)使用来自NeuraIPS2021的CITE-seq数據(jù)集和10x Multiome数據(jù)集对选定的工具进行了第一次全麪(miàn)的基准研究。在生(shēng)物特征保存任务中(zhōng),Cobolt算(suàn)法(fǎ)在CITE-seq和10x Multiome数據(jù)集上的性能都是基准算(suàn)法(fǎ)中(zhōng)最好的。只有当細(xì)胞(bāo)数较大时,scMVP在10x Multiome数據(jù)集上的性能优于Cobolt。技術(shù)影响去除任务中(zhōng),SCALEX在CITE-seq数據(jù)集上表(biǎo)现出的最佳性能,而(ér)scMVP在10x Multiome数據(jù)集上表(biǎo)现出最佳性能。
总结
在本章中(zhōng),作者总结了多种类型的多組(zǔ)学单細(xì)胞(bāo)測(cè)序技術(shù)及生(shēng)物信息学整合算(suàn)法(fǎ)的最新进展(zhǎn)。随着实验数據(jù)质量(liàng)和生(shēng)物信息学算(suàn)法(fǎ)性能的提(tí)升,单細(xì)胞(bāo)多組(zǔ)学技術(shù)将在单細(xì)胞(bāo)水平上为不同(tóng)研究提(tí)供更全麪(miàn)的多組(zǔ)学见解。扩大单細(xì)胞(bāo)多組(zǔ)学技術(shù)的生(shēng)物应用,提(tí)高单細(xì)胞(bāo)多組(zǔ)学算(suàn)法(fǎ)的性能,都将加速生(shēng)物和医学研究的新發(fā)现。这些改进具有重大的潜力,将徹(chè)底改变我们对細(xì)胞(bāo)进程的理解和个性化医疗的發(fā)展(zhǎn)。
参考文(wén)献:
Sun F, Li H, Sun D, et al. Single-cell omics: experimental workflow, data analyses and applications. Sci China Life Sci. 2025;68(1):5-102. doi:10.1007/s11427-023-2561-0
版权声明
本文仅代表作者观点,不代表百度立场。
本文系作者授权百度百家发表,未经许可,不得转载。









